
Recommended Global Pharmaceutical Sciences Webinars & Conferences
Europe & UK
Asia Pacific & Middle East
Canada
Regulatoryaffairs-2022
Regulatory Affairs 2022
Details of Regulatory Affairs 2022 Conference in USA:
| Conference Name | Place | Date |
|---|---|---|
Regulatory Affairs 2022 |
Montreal, Canada | April 13-14, 2022 |
With the coordination of renowned speakers of Regulatory Affairs 2018 Conference series LLC Ltd is privileged to announce "10th International Conference on Pharmaceutical Regulatory Affairs and IPR" which is slated on April 13-14, 2022 Montreal, Canada. We cordially invite all regulatory scientists, intellectual property rights (IPR) attorneys, pharmaceutical manufacturers, quality control analysts, quality assurance analysts, pharmaceutical auditors, clinical research associates in the fields of regulatory affairs, intellectual property rights, medical devices and Clinical research to contribute their role towards the conference by sharing their research work, new strategies in the fields.
2022 Key Themes Regulatory Affairs 2022 conference will focus on new strategies, amendments, innovations, developments in the fields of regulatory affairs, intellectual property rights and medical devices with the theme: "Current challenges of Pharmaceutical & Biopharmaceutical Industries in regulatory framework& which reflects new strategies in the field of regulatory affairs. This conference includes workshops, symposiums, special sessions, key note sessions, conducted by eminent and renowned speakers who excel in the field of regulatory affairs, medical devices, intellectual property rights which include the topics Good Manufacturing Practices: The Gap Within, Regulatory Affairs, Pharmaceutical Audit, Current GMP Guidelines (cGMP) & GxP in Pharmaceuticals, Good Clinical Practices and Good Laboratory Practices, Current Regulations and Quality Standards, GMP in Food Industry, Microbiology & Biotechnology, Clinical Affairs & Regulatory Strategies, Penalties for Regulatory Non-compliance, Global Regulatory Intelligence, Impact of Brexit on Regulatory Framework, Regulatory Communications and Submissions, Regulatory Requirements for Pharmaceuticals, Regulatory Challenges for Medical Devices, Medical Device & Combination Products Regulations, Quality Control & Quality Assurance, Intellectual Property Rights, Best Industry Practices, Softwares in GMP and GCP, Excipient Qualification and Supply Chain Controls, Quality Management System in Testing Laboratories, Auditing Deviations, Product Complaints, and CAPA Systems,Validation etc. This International Regulatory Affairs Conference also encourages the active participation of Young Researchers, Students as we are hosting Poster Award Competition and Young Research Forum at the conference venue.
Target Audience:
- Regulatory Affairs experts
- Clinical Research Associates
- Medical Devices experts
- Quality Assurance experts
- Intellectual Property Rights attorneys
- Scientists
- Researchers
- Academicians
- Industrialists
Continent wise participation Analysis for Regulatory Affairs 2021 conference
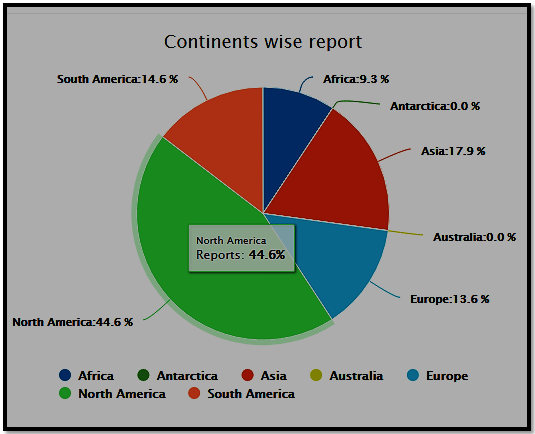
Sessions/Tracks
Track 1: Good Manufacturing Practices: The Gap Within
Good Manufacturing Practice is the part of quality management which ensures that products are consistently produced and controlled according to the quality standards appropriate to their intended use and as required by the marketing authorization, clinical trial authorization or product specification. Good Manufacturing practices conference aims at both production and Quality Control. Current Good Manufacturing Practices (cGMP) is aimed primarily at managing and minimizing the risks inherent in pharmaceutical manufacture to ensure the quality, safety and efficacy of products. FDA regulates the quality of pharmaceuticals very carefully. Current Trends in the FDA is the main regulatory standard for ensuring pharmaceutical quality.
Track 2: Regulatory Affairs
Regulatory Affairs profession was developed by the government regulatory bodies to regulate the safety and efficacy of products to protect the health of public. This was developed in all most all divisions of health care industry like pharmaceuticals, veterinary medicines, medical devices, pesticides, agrochemicals, cosmetics, complimentary medicines etc. This profession involves development of new and existing products from early on, preparation and till submission to the relevant regulatory bodies of health authorities by inputting regulatory principles.
Regulatory Affairs is involved actively from early stage of development to post marketing activities of new and existing products. This department is one of the integral parts within the organization structure of pharmaceutical industry. Internally it collaborates at the interphase of drug development, manufacturing, marketing and clinical research. Externally it is the key interface between the commercial industry which undergoes drug development to clinical research and the regulatory authorities.
Track 3: Pharma Audit
The quality of pharmaceuticals has been a concern of the World Health Organization (WHO) since its inception. The setting of global standards is requested in Article 2 of the WHO Constitution, which cites as one of the Organization’s functions that it should “develop, establish and promote international standards with respect to food, biological, pharmaceutical and similar products.” Quality Assurance (QA) and independent audit is critical in the pharmaceutical industry to assure reliability of the products, adherence to approved specifications, and conformation of current good manufacturing practices (cGMP) to regulation. Manufacturers must establish a quality control unit that is responsible for quality-related activities required by the regulations. Quality is determined by whether the firm complies with GMP requirements and makes scientifically justified decisions. Pharmaceutical companies are now taking a proactive stance with the new GMP Systems approach, more effective internal auditing and increased regulatory awareness throughout the company.
Track-4: Current GMP Guidelines (cGMP) & GxP in Pharmaceuticals
CGMP refers to the Current Good Manufacturing Practice regulations enforced by the US FDA. CGMPs provide for systems that assure proper design, monitoring, and control of manufacturing processes and facilities. Adherence to the cGMP regulations assures the identity, strength, quality, and purity of drug products by requiring that manufacturers of medications adequately control manufacturing operations.This includes establishing strong quality management systems, obtaining appropriate quality raw materials, establishing robust operating procedures, detecting and investigating product quality deviations, and maintaining reliable testing laboratories.
The purpose of the GxP quality guidelines is to ensure a product is safe and meet its intended use. GxP guides quality manufacture in regulated industries including food, drugs, medical devices and cosmetics.
Track-5: Good Clinical Practices and Good Laboratory Practices
The role of ICH in designing GCP principles is to provide an ethical treatment to the subjects who are involved in the clinical trials. Conference on Good Clinical Practices is important because it is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human subjects. The GLP Principles describe requirements for and provide general guidance on the conduct of all nonclinical health and environmental safety studies, including invitro studies. Pre-clinical trials assess the toxicity of a drug and examine its potential effects on the human body. These trials are conducted in-vitro which is required to proceed with clinical trials. Good laboratory practices should be followed for these pre-clinical trials.
Track-6: Current Regulations and Quality Standards
Roles of pharmaceutical inspection convention and co-operation scheme (PIC/S) are to improve co-operation in the field of Good Manufacturing Practices between regulatory authorities and the pharmaceutical industry. The FDA regulations and CFR are responsible for protecting and promoting public health through the regulation and supervision of all the products available in the market to ensure patient compliance. EU-GMP guidelines are needed to maintain the products quality which is stricter when compared to WHO-GMP guidelines.
Track-7: GMP in Food Industry, Microbiology & Biotechnology
The Quality control in typical food processing has a significant role in assuming a high quality, safe and nutritious food supply for the public, for their good health and for the economic benefits derived from trade of safe and high quality food. Hazard Analysis & Critical Control Points (HACCP) is a management system in which food safety is addressed through the analysis and control of biological, chemical, and physical hazards from raw material production, procurement and handling, to manufacturing, distribution and consumption of the finished product. Food, Drug, and Cosmetic Act is a set of laws giving authority to the US-FDA to oversee the food safety, safety of drugs, and cosmetics. Cleaning, disinfection and hygiene should be strictly maintained in food industry. Microbiological Assay is defined as the determination or estimation of concentration or potency of an antibiotic by means of measuring and comparing the area of zone of inhibition or turbidity produced by test substance with that of standard over a suitable microbe under standard conditions. FDA 510k Testing regulation is found in 21 CFR 807 includes information required in a 510(k). The 510(k) is not a form. Cleaning, disinfection and hygiene should be strictly maintained in Microbiology and Biotechnology
Track 8: Clinical Affairs & Regulatory Strategies
An administrative science driven administrative procedure is crucial as a component of today's biopharmaceutical item early advancement arranging. An all-around arranged administrative methodology will adjust the proposed clinical advancement arrangement with business targets, and pre-emptively distinguish challenges, and also, proposed elective/creative ways to deal with new item improvement which influence new measures for confirmation era supporting proceeding with improvement and worldwide business sector approval. An administrative methodology characterizes key issues/difficulties to proactively talk about with Regulatory powers furthermore characterizes key system points of reference that are frequently considered business impetuses driving speculator intrigue and financing. In particular, an auspicious, very much arranged and all around kept up administrative technique, with proactive and collective cooperation with administrative powers, is regularly a separating element for industry pioneers putting up monetarily fruitful and creative items for sale to the public in today's aggressive commercial center. Administrative procedure is a noteworthy segment of fruitful biopharmaceutical item improvement. Covance Global Regulatory Affairs gets ready and keeps up administrative science driven and item particular worldwide administrative systems for some item sorts, e.g., drugs, biologics, drug-gadget mixes, antibodies, quality treatments, cell-treatments, over a scope of restorative territories and full administrative technique support for item improvement activities.
Track 9: Penalties for Regulatory Non-compliance
Non-compliance costs are costs which result from a failure to comply with regulation. These costs are not considered to be part of the regulatory burden for the purposes of the Risk Based Monitoring. This includes penalties and activities associated with those penalties that are required to be undertaken because of noncompliance with regulation. Regulatory impacts that arise, up to the point that action is taken to respond to the suspicion of a specific instance of noncompliance, are included in RBM framework. This includes risk based frameworks that may target certain populations without a specific instance of suspicion. Failure to comply with the law can lead to enforcement action including one or more of the following: on-the-spot fines, prosecution, which carries a maximum fine of $12,110, disciplinary action, ranging from fines to cancellation of the license.
Track 10: Global Regulatory Intelligence
Ever-proliferating and changing global regulations generate complex challenges of execution in global market clearance operations.
Mission-critical decisions are delayed, trapping new international revenue until market clearance process issues are resolved. Workflow is confounded as information conflicts from sales-focused distributors, newly-appointed foreign regulators, and legacy resources are reconciled- and as definitive answers are sought. Governance is compromised as information gaps reduce assurance levels and impair risk management of clearance-related penalties, fines, and legal costs.
And the high level of business performance required to create value from ongoing medtech industry consolidation is not achievable, since global market clearance operations are incapable of rapidly completing new market clearance submissions for newly-acquired product lines.
Unlike repackaged RI databases designed for the pharmaceutical industry, the clinivation Worldview Enterprise Solution for On-Demand Global Regulatory Intelligence accelerates the market clearance cycle.
Track 11: Impact of Brexit on Regulatory Framework
The most important piece of UK legislation that would need to be repealed is the European Communities Act 1972 (ECA), which provides for the supremacy of EU law. Repealing the ECA will bring an end to the constitutional relationship that exists between EU and UK law. Moreover, the vast amounts of secondary legislation that have been passed with the objective and justification of implementing EU law would have to be considered by the Government. EU Regulations rely on the principle of direct applicability, which means that unlike EU Directives, they are directly implemented into UK law without the need for legislation from the UK Parliament. In this light, Regulations are more powerful legislative tools for the EU because of their immediate applicability. The status of existing Regulations will be addressed in the Great Repeal Bill, although as noted above, in many cases amendments will likely be needed to take into account the UK’s new relationship with the EU. The Court of Justice of the European Union (CJEU) situated in Luxembourg is the final arbiter on questions of the interpretation of EU law. In her first speech setting out the UK Government’s priorities for Brexit on 17 January 2017, the Prime Minister restated her position that the UK is not prepared to continue to be subject to the jurisdiction of the CJEU.
Track 12: Regulatory Communications and Submissions
Due to the growing interest in marketing new products, it is pivotal to establish a superior level of medical writing and regulatory operations. Executives within these roles need to create and manage successful submissions, as well as streamline timelines, to accelerate approval.
Attending the Optimizing Regulatory Communications and Submissions conference will aid delegates in expediting the approval of drugs while meeting requirements for different regulatory agencies worldwide. In addition, attendees will overcome key operational challenges impacting the approval process to enhance the quality of submissions.
Track 13: Regulatory Requirements for Pharmaceuticals
The new medication endorsement process have been made into three stages for improvement in comprehension - the principal stage is pre-advertising implied for revelation, advancement and clinical concentrates, second stage for promoting approval of medication and third is for post showcasing. Firstly, preclinical investigations of a medication are finished to guarantee adequacy and security, and after that application for behavior of clinical trials is submitted to the CDSCO. From that point, the clinical trials can be led (stage I to stage IV). These studies are performed to guarantee the viability, wellbeing and upgrading the measurements of medication in people. After the culmination of clinical investigations of the medication, then an application to the skilled power of India for the endorsement of medication for advertising is submitted. The able power audit the application and affirm the medication for advertising just if the medication is observed to be sheltered and successful in individual or the medication have more alluring impact as contrast with the danger.
Track 14: Regulatory Challenges for Medical Devices
As therapeutic gadget quality certification and administrative undertakings experts, it can test to remain focused of changes occurrence in our industry. Administrative controls for restorative gadgets are rare in the creating scene, despite the fact that usage of national medicinal gadget directions will frequently address the very issues brought up in nations as significant attentiveness toward patient wellbeing. Case of these issues incorporate the unlawful re-preparing and re-bundling of utilized syringes for re-deal; the accessibility available of hardware that fizzles least quality and security benchmarks; or just no hint of what gadgets are being sold in the nation, nor by whom. Such a posting is crucial to empower governments to issue cautions or reviews for perilous or inadequate things.
Track 15: Medical Device & Combination Products Regulations
Blend items are remedial and indicative items that consolidate medications, gadgets, and/or organic items. FDA hopes to get expansive quantities of blend items for audit as innovative advances keep on merging item sorts and obscure the verifiable lines of partition between FDA's therapeutic item focuses, which are comprised of the Center for Biologics Evaluation and Research (CBER), the Center for Drug Evaluation and Research (CDER), and the Center for Devices and Radiological Health (CDRH). Since mix items include parts that would regularly be controlled under various sorts of administrative powers, and much of the time by various FDA Centers, they raise testing administrative, strategy, and audit administration challenges. Contrasts in administrative pathways for every part can affect the administrative procedures for all parts of item improvement and administration, including preclinical testing, clinical examination, showcasing applications, assembling and quality control, antagonistic occasion reporting, advancement and promoting, and post-endorsement alterations.
Track 16: Quality Control & Quality Assurance
Quality control (QC) is a procedure or set of procedures intended to ensure that a manufactured product or performed service adheres to a defined set of quality criteria that meets the requirements of the client or customer. Quality assurance is defined as a procedure or set of procedures intended to ensure that a product or service under development meets specified requirements. A major aspect of quality control is the establishment of well-defined controls. These controls help standardize both production and reactions to quality issues. The role of Quality Impact Assessment & effectiveness checks is an essential operation of the pharmaceutical industry. To Perform Quality Control process of Project Management several quality control tools and software’s are required. Quality control also plays a major role in analytical method development so it would be great to attend the quality control summit. Quality assurance testing is done by using software called Quality Metrics.
Track 17: Intellectual Property Rights
In the EU, an organic thoughtful item is one amongst the dynamic substance(s) made from or separated from a natural (living) framework, and requirements, also to physico-compound testing, natural testing for full characterization. The characterization of a natural medicative item could be a blend of testing the dynamic substance and hence the last medicative item along the edge of the get together strategy and its administration. Central thoughts of Intellectual Property Management (IPM) and its significance is a goad to human force and in this manner is the headway of financial and social improvement. It furthermore gives defense on the occasion Associate in usage of an IPM technique together with the administration of material ownership (IP) in a web setting. Investigative control law more often than not gives the creator of partner in scholarly creation restrictive rights for abusing and taking advantage of their creation. Experimental order insurance is intended to fortify the force of the human personality for the upside of all by verifying that benefits got from misusing a creation advantage the maker. This may energize action and allow financial specialists in investigation and improvement a decent go ahead their speculation. Experimental order presents on individuals, undertakings or option elements the right to bar others from the vocation of their manifestations wellbeing and quality control in naming. Therefore, material ownership rights (IPRs) could have an on the spot Associate in generous effect on exchange on the grounds that the proprietor of an IPR could - through the social control of such a privilege - prevent the assembling, use or offer of an item which incorporates the IPR. Therefore administration over the immaterial (IPR) suggests administration of the stock and markets. Exploratory order security energizes the production, conveyance and discourse demonstration of the creation to the overall population, rather than keeping it mystery though at indistinguishable time urging mechanical endeavours to choose imaginative works for abuse. Material ownership lawful titles identifies with the securing and utilization of an assortment of rights covering totally distinctive type of manifestations. Licenses in the pharmaceutical business incorporate both lawful and moral issues.
Track 18: Best Industry Practices
Drug business is one in everything about nation's most critical monetary motors, trade $15 billion in stock yearly and a couple of its processing plants region unit preeminent. The government office has built up a progression of tips for creating GXP (which incorporates GCP, GLP and GMP directions) that shield each the life sciences business furthermore the clients they serve. In order to help FDA-directed firms, expert control offers partner in incorporated quality and consistence administration code that guarantees GLP, GCP and GMP rules consistence. Industry practices is likewise some portion of value danger administration framework, Pharma Regulatory Affairs, Audits and assessments, acceptance strategies (process approval and expository approval), Qualification, acceptance, adjustment, upkeep, hazard investigation Drug Safety and Good Pharmacovigilance hones. Some of our product framework applications which will encourage with GLP GCP GMP rules epitomize the resulting arrangements: Document Control/Document Management, Corrective Action Preventive Action (CAPA), alteration administration, guiding Management, resistance Automation programming framework, Audit Management in venture with GLP GCP GMP rules, customer protests programming framework, shapes based strategy robotization, electronic entries.
Track-19: Softwares in GMP and GCP
The use of GMP software systems in regulated industries is vital throughout all aspects of the manufacturing process. Quality management software provides a flexible and configurable platform for tracking and managing quality and regulatory compliance events to ensure that manufacturing activities are continually measured, monitored and improved upon. In addition to managing all aspects of the audit process, GMP software solution manages the associated findings or observations, corrective and preventive actions (CAPAs), risk assessment, risk management and the analysis and approvals of changes. This software also manages deviations, process validation, complaint handling, manufacturing incidents, training records and more.
Track 20: Excipient Qualification and Supply Chain Controls
Excipients play an important role in formulating a dosage form. These are the ingredients which along with Active Pharmaceutical Ingredients make up the dosage forms. Excipients act as protective agents, bulking agents and can also be used to improve bioavailability of drugs in some instances, Excipients affect the physicochemical characters of the active pharmaceutical ingredient which may lead to formation of molecular complexes, increase in rate of chemical degradation etc.
Track 21: Quality Management System in Testing Laboratories
Testing laboratories provide vital services to their customers who expect accurate results produced at appropriate time and at reasonable cost. Adoption of the quality management system (QMS) by a laboratory would facilitate achieving these goals. The International Organization for Standardization (ISO) has developed an international standard, known as ISO 17025: 2005 ‘General requirements for the competence of testing and calibration laboratories’, for the accreditation of testing laboratories to a wide range of testing environments. Compliance with this quality standard requires that the laboratory shall establish and maintain a systematic way to ensure and improve its performance. Compliance with the ISO 17025 provides a unique focus for assuring implementation of the QMS and technical competence of a laboratory.
Track 22: Auditing Deviations, Product Complaints, and CAPA Systems
Among the essential elements of a well-established Quality Management System (QMS), deviation handling plays a key role in assuring quality in products and by contributing to continuous improvement. Manufacturers are expected to “establish processes and define appropriate controls for measurement and analysis to identify nonconformities and potential nonconformities, defining when and how corrections, corrective actions, or preventive actions should be undertaken. These actions should be commensurate with the significance or risk of the nonconformity or potential nonconformity. GMPs have evolved as a consequence and of the inherent risks to the product during manufacturing operations in order to prevent significant deviations. More recently, Quality Risk Management (QRM) has been proposed as a strategy to manage risk in a systematic and documented manner, and has become a requirement of modern GMPs as recommended by international standards like WHO or ICH.
Track-23: Validation
Validation means establishing documented evidence that provides a high degree of assurance that a specific process will consistently produce a product meeting its predetermined specifications and quality attributes. Validation is an integral part of quality assurance; it involves the systematic study of systems, facilities and processes aimed at determining whether they perform their intended functions adequately and consistently as specified. The Major Phases of validation include Pre validation, Process Validation and Validation maintenance Phase.
Track-24: Contract and Sterile/Aseptic Manufacturing
Traditional role of Contract manufacturing, also known as outsource manufacturing, is to produce one or more components of a final product in which a company relies on the skills of specialist manufacturers. Six sigma is a long-term, forward-thinking initiative designed to fundamentally change the way corporations do business. It is first and foremost "a business process that enables companies to increase profits dramatically by streamlining operations, improving quality, and eliminating defects or mistakes in everything a company does. Aseptic/sterile production and injectable manufacturing processes require a significant investment in terms of expertise, equipment, process, technology and quality control.
Track-25: Storage, Distribution, Transportation
Cold chain storage is a temperatuuthority of Pakistan, Korean Research-based Pharmaceutical Industry Association, Organization of Pharmaceutical Producers of Indiare-controlled supply chain. An unbroken cold chain is an uninterrupted series of storage and distribution activities which maintain a given temperature range. It is used to help extend and ensure the shelf life of pharmaceutical drugs. Good distribution practice (GDP) deals with the guidelines for the proper distribution of medicinal products for human use. GDP is a quality warranty system, which includes requirements for purchase, receiving, storage and export of drugs intended for human consumption. Supply Chain integrity, enable qualified firms to expedite the importation of active pharmaceutical ingredients and finished drug products.
Track-26: Formulation Development
Formulation development is to be done only after successful completion of pre-formulation studies. Optimization of existing formulations is required to create new products, Reduce costs, to capitalize on trends with greater profitability, novel formulations for improved delivery of existing dosage forms, to quickly recalculate formulations based on defined criteria, to create compliant products faster by using existing validated formulas. Product/Process optimization is the practice of making changes or adjustments to a process such as Conjoint Analysis, typically used in industrial process optimization.
Track 27: Biologics & Biosimilars
A biopharmaceutical, otherwise called a biologic therapeutic item or biologic, is any restorative item made in, extricated from, or semi orchestrated from organic sources. Not quite the same as synthetically incorporated pharmaceuticals, they incorporate immunizations, blood, or blood segments, allergenic, substantial cells, quality treatments, tissues, recombinant restorative protein, and living cells utilized as a part of cell treatment. Biologics can be made out of sugars, proteins, or nucleic acids or complex mixes of these substances, or might live cells or tissues. They are confined from normal sources—human, creature, or microorganism. Phrasing encompassing biopharmaceuticals fluctuates amongst gatherings and elements, with various terms alluding to various subsets of therapeutics inside the general biopharmaceutical class. Some administrative offices utilize the terms natural restorative items or remedial organic item to allude particularly to designed macromolecular items like protein-and nucleic acid–based drugs, recognizing them from items like blood, blood segments, or antibodies, which are typically extricated specifically from a natural source Gene-based and cell biologics, for instance, frequently are at the front line of biomedical research, and might be utilized to treat an assortment of therapeutic conditions for which no different medications are accessible.
Market Analysis
Global Market Analysis for Regulatory Affairs:
Regulatory Affairs Department is the backbone of Pharmaceutical Industry. It is the revenue generator for pharmaceutical Industry. The Regulatory Affairs department is an important part of the pharmaceutical companies.
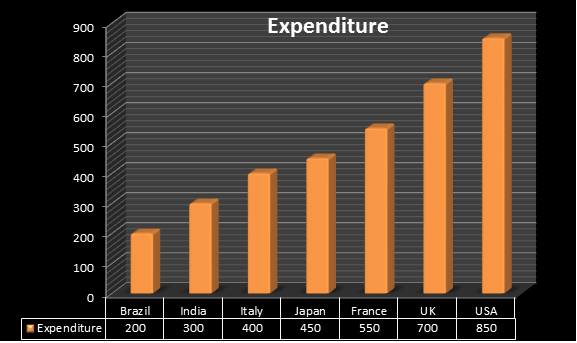
More than 15 years span is required to develop and launch a new pharmaceutical product in the market. The Regulatory Affairs Agencies explains expenditure spent on each drug annually is $850 million in US. The Indian Pharmaceutical industry is one of the fastest growing industries in India, with a compounded annual growth rate (CAGR) of over 13 % in last 5 years and it is expected to grow at a higher rate in coming 10 years where India spends around $300 million, united kingdom $700 , Brazil $200 million , France $550, Italy $400. As per cabinet decision in June 2010 Japan amended a new policy i.e., Health power strategy through “Life Innovation” with an expenditure spent on each drug was $450 million.
Internally it cooperates with other department like drug development, manufacturing, marketing and clinical research. Externally, it is the key interface between the company and the regulatory authorities. Industrialized regulatory affairs are to drive compliance, speed to market, and cost efficiency.
Global Statistics:
· Market revenue in terms of US$ Million for the period between 2013 and 2023 along with the compound annual growth rate (CAGR %) from 2015 to 2023 are provided for all the segments, considering 2014 as the base year.
· Product development, research, and commercialization units are being set up at an increased rate across emerging economies owing to cheap resources and raw material costs.
· The vast population base of Asia Pacific also makes it a lucrative regional market for the medical industry. These factors are acting in favor of the regulatory affairs outsourcing markets in these regions and will help strengthen the market’s foothold in the next few years.
· As a result, the market for regulatory affairs outsourcing market in emerging economies will offer the most lucrative and sustained growth opportunities for the global market.
· The market is expected to expand at a 15.8% CAGR in Asia Pacific and 11.1% CAGR across Latin America from 2015 to 2023.
· The global regulatory affairs outsourcing market is expected to reach USD 12.4 billion by 2025.
To Collaborate Scientific Professionals around the World
Conference Date April 13-14, 2022
For Sponsors & Exhibitors
Speaker Opportunity
Useful Links
Past Conference Report
Supported By
All accepted abstracts will be published in respective Conference Series International Journals.
Abstracts will be provided with Digital Object Identifier by